
|
Nauka » Biologia » Biologia molekularna Priony. Czyżby nowożytny miecz Damoklesa? [3] Przybranie innej formy przestrzennej przez białko prionowe jest jednoznaczne z nabyciem przez niego właściwości białka infekcyjnego (patogennego) [ 28 ]! Znamienne, że wciąż nie znane są przyczyny zmian PrP w postaci patogennej. 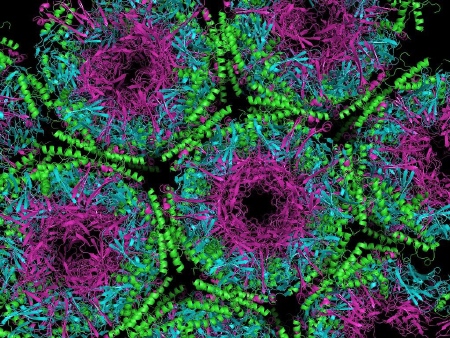 Struktura krystalograficzna białka prionowego [ 29 ] Zmiana kształtu jest przekazywana kolejnym cząstkom białka. Specyfika polega na tym, że nie jest to typowe powielanie, namnożenie, lecz przekazanie zmian czysto strukturalnych innym, występującym już na obszarze komórki cząstkom pokrewnym, zostają one niejako zmuszone do przyjęcia nieprawidłowej konformacji przestrzennej. Zmienione priony są odporne na działanie enzymów (proteaz), nie mogą zostać strawione, akumulują się więc wewnątrz neuronów (tworzą płytki amyloidu, tzw. pałeczki prionowe [ 30 ]) i zlepiają w charakterystyczne dla poszczególnych szczepów złogi, uszkadzając ciało komórki (wakuolizacja) oraz nabierając cech neurotoksycznych. Konsekwencją są ubytki tkanki mózgowej, stąd też nazwa gąbczasta encefalopatia. Postępująca degeneracja powoduje ciężkie zaburzenia psychiczno-ruchowe, jak dotąd nieuchronnie prowadząc do śmierci. Obecność patogennych prionów, które są wszak w normalnej postaci cząstkami słabo immunogennymi, nie powoduje wytworzenia wykrywalnych ilości przeciwciał we krwi, czy płynie mózgowo-rdzeniowym (stąd trudności w diagnostyce), są one jednak komercyjnie dostępne (wytworzone laboratoryjnie). Wszystkie choroby prionowe ludzi i zwierząt charakteryzują się zmianami zwyrodnieniowymi układu nerwowego, dlatego określane są wspólnym skrótem TSE (transmissible neurodegenerative disease = pasażowalne encefalopatie gąbczaste). Przyjęto, że choroby te charakteryzują się gąbczastą patologią oraz obecnością zgrupowań kryształów PrP (widoczne pod mikroskopem elektronowym jako skręcone włókienka w homogenizowanym preparacie tkanki mózgowej) zwanych SAF (scrapie associated fibrils). 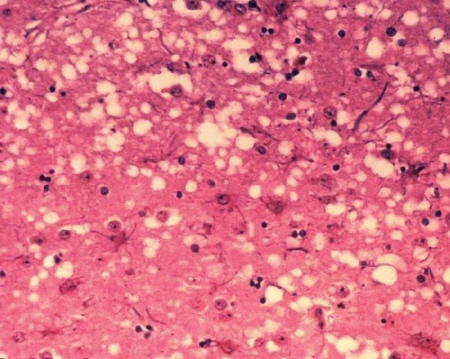 Mikrofotografia tkanki mózgowej ujawniająca zmiany cytoarchitektoniczne w gąbczastej encefalopatii bydła. Obecność wakuol („dziur") w istocie szarej daje gąbczasty wygląd tkanki [ 31 ] W przeciwieństwie do innych czynników patogennych jak bakterie, wirusy, priony nie są obce względem atakowanego organizmu, lecz stanowią jego naturalne elementy strukturalne! Zmiana fenotypu związana z prionami coraz częściej uznawana jest za formę informacji genetycznej, mimo, że w jej przekazywaniu nie pośredniczą kwasy nukleinowe (dotyczy jedynie organizmów haploidalnych, rozmnażających się przez podział). Z tego punktu widzenia badacze zaliczają ją do zjawiska epigenetycznego, podobnie jak tzw. kod histonowy, czyli modyfikacje posttranslacyjne histonów powodujących rozmaite zmiany nie tylko w stanie chromatyny, a także komórki. [ 32 ] Przy badaniach związanych z prionami obowiązują precyzyjnie wyznaczone procedury: poziom bezpieczeństwa P2 (śluza, fartuch, czapka itp.) do P3 (dodatkowo tworzy się warunki ujemnego ciśnienia). Cząstki patogennych prionów są zdumiewająco odporne, w warunkach tzw. suchego spalania ulegają zniszczeniu dopiero w temperaturze +360ºC [ 33 ] lub, aż +600ºC. [ 34 ] Przy podwyższonym ciśnieniu do spalenia wystarcza temperatura około +135ºC. [ 35 ] Czynnik scrapie badał (lata 1939-1953) D. R. Wilson, jeden z jego kolegów pisał: „(...) czynnik chorobotwórczy jest niezwykle odporny na wszelkie substancje niszczące. Przetrzymał półgodzinne gotowanie. W zamrażarce przetrwał dwa miesiące. Był nieczuły na środki dezynfekcyjne, zawierające silne stężenie formaldehydu, fenolu i chloroformu. Przenikał przez najgęstsze filtry i pozostał w roztworze umieszczonym w wirówce przy czterech tysiącach obrotów na minutę. Mógł przeżyć w wysuszonym mózgu przez co najmniej dwa lata, był odporny na silną dawkę promieniowania ultrafioletowego. Przenosił się z jednej owcy na drugą wstrzyknięty podskórnie, domięśniowo czy bezpośrednio do mózgu." [ 36 ] Naukowcy postanowili sprawdzić jak priony działają na białko obce naturze, stworzone przez człowieka, w tym przypadku połączono białka dwóch drożdży: "SC" (Saccharomyces cerevisiae) i "CA" (Candida albicans). Wynik: białko okazało się podatne zarówno na działanie oryginalnych prionów "SC", jak i "CA", tym samym właściwie było podatne na działanie prionów obcych sobie gatunków. Możliwe więc iż podatna na zmiany nie musi być cała cząsteczka, lecz wystarczy by jej fragment był wrażliwy na czynnik chorobotwórczy, a dojdzie do przeniesienia choroby. Do dziś poznano następujące choroby prionowe człowieka, wszystkie są neurodegeneracyjne, powstają w obrębie ośrodkowego układu nerwowego. Wszystkie również — jak dotąd — zawsze kończą się śmiercią, leczenie jest wyłącznie objawowe! Nie jest znane lekarstwo na choroby prionowe. Choroby prionowe człowieka: KURU Chociaż dzieje ludzkich chorób prionowych zaczęły się właśnie od niej dla przeciętnego człowieka choroba ta pozostaje właściwie nieznana. Wynika to po części z faktu, iż ma (miała) charakter lokalny, występując wyłącznie w obrębie plemienia Fore, mieszkańców górzystych rejonów Papui-Nowej Gwinei („kuru" oznacza w języku Fore „drżeć z zimna/gorączki"). Praktykowali oni rytualny kanibalizm w dowód szacunku dla zmarłych. [ 37 ] Okres zwiastunów (prodromalny) — trwa od pojawienia się pierwszych objawów do pełnego rozwoju objawów klinicznych — obejmuje: bóle głowy, brzucha i kończyn (stawów), utratę masy ciała. Objawy (występowały po okresie kilkunastu miesięcy lub nawet 40 lat): 1. Zawsze występuje śmiertelna ataksja móżdżkowa [ 38 ], której towarzyszą drżenie, ruchy mimowolne o typie pląsawiczych/atetotycznych oraz nietrzymanie moczu i kału. 2. Kuru ma trzy fazy: — chory jeszcze chodzi — chód staje się niepewny, przechodzi w nasiloną ataksję i abazję [ 39 ]. Pojawia się delikatne „drżenie z zimna", potem chwianie, próby utrzymania równowagi (kurczowe przytrzymywanie się gruntu za pomocą szponowato zgiętych palców stóp). Może pojawić się poziomy oczopląs. Odruch podeszwowy zawsze zgięciowy. [ 40 ] Zawsze obecny klonus (trząs) rzepki, ewentualnie klonus stopy, czyli ciąg mimowolnych skurczów włókien mięśniowych wywołanych przez nagłe rozciąganie mięśnia. — chory może siedzieć — zaczyna się, gdy chory nie jest w stanie chodzić bez trwałego podparcia (koniec przychodzi z chwilą, gdy nie potrafi siedzieć bez podparcia). Postępowanie objawów: niestabilność postawy, nasilona ataksja, grubofaliste drżenie, dyzartria (jeden z typów zaburzeń mowy, wynikający z dysfunkcji aparatu wykonawczego: języka, podniebienia, gardła, krtani). — terminalna — osoba jest już całkowicie unieruchomiona. Nie trzyma moczu i kału. Pojawia się ciężka dysfazja (zaburzenia mowy, brak koordynacji słów i trudności ułożenia ich w zdania mimo sprawności odpowiednich mięśni). Specyficzny dla kuru jest brak otępienia (lub też pojawia się ono w bardzo zaawansowanych stadiach). Występują prymitywne odruchy: ssania, gryzienia, chwytny. Zmiany nastroju, od euforii do depresji. Następnie przymusowy płacz i śmiech (tzw. „patologiczny śmiech", stąd choroba zwana jest „śmiejącą się śmiercią"), otępienie (demencja). Ostatecznie wyniszczeni i niezdolni do ruchu chorzy oczekiwali śmierci głodowej lub ginęli wskutek zachłystowego zapalenia płuc, jeżeli podano im odrobinę wody. Choroba zaczęła zanikać od lat 40. XX w. kiedy to rząd ostatecznie wyegzekwował zakaz rytualnego spożywania mózgów zmarłych współplemieńców. Obecnie kuru nie występuje. [ 41 ]
Przypisy: [ 28 ] Obala
to zresztą pokutujący w biologii dogmat według którego kolejność aminokwasów
determinuje strukturę przestrzenną cząstki białka. Tymczasem białka mogą mieć tę
samą sekwencję aminokwasów, przy innej strukturze. Pierwszym, który wykazał, że
PrPc ma inną konformację niż PrPSc był Byron Caughey z Montana Rocky Laboratory (USA). 12 lipca 1999 r. uczeni z Uniwersytetu
Kalifornijskiego w San Francisco przenieśli priony PrPSc w czystej postaci do komórek drożdży i wykazali, że ich obecność wywołuje
przekształcenia nieszkodliwych białek. [ 29 ] źródło:
University of Liverpool [w:]
http://www.stfc.ac.uk/ [ 30 ] Twory
te odkrył w tkankach (pod mikroskopem optycznym) już w XIX w. Robert Koch;
przypomniały mu one ziarna skrobi w miąższu gruszki stąd nazwa amyloid (z łac.
amylum — skrobia). [ 31 ] źródło:
http://pl.wikipedia.org/wiki/ [ 32 ] Epigenetyka — badanie dziedziczności pozagenowej. Histony
(białka
histonowe), to małe zasadowe białka łączące się z cząsteczkami DNA, są
podstawowym składnikiem białkowym chromatyny. Wyróżnia się cztery rodziny
histonów: H2A, H2B,H3, H4 i H1. [ 33 ] Dane z 1995 r. [ 34 ] Dane z 2000 r. [ 35 ] Istnieją
podejrzenia, że priony zachowują aktywność nawet po zadziałaniu temperatury
przekraczającej +200ºC. Inne źródła: przy podwyższonej temperaturze (gotowanie,
pieczenie) wokół cząsteczki prionu tworzy się ochronna powłoka. By ją zniszczyć,
należy zwiększyć ciśnienie lub zastosować podgrzewanie do minimum +600ºC. [ 36 ] [w:]
R. Rhodes, op. cit., s.97. [ 37 ] Zwyczaje
te Fore przejęli od sąsiednich plemion dopiero na przełomie wieków XIX i XX.,
następnie endokanibalizm (kanibalizm w obrębie rodziny) rozprzestrzenił się na
północ. Epidemia kuru wyraźnie zaburzyła proporcje płci, dotyczyła niemal
wyłącznie kobiet i dzieci. Mężczyźni chorowali rzadko, stąd też ich liczba w południowym Fore była dwukrotnie wyższa niż kobiet (w niektórych wioskach
stosunek wynosił 3:1). Według samych Fore, kuru było efektem czarnoksięstwa -
czarnoksiężnika brutalnie zabijano podczas obrzędu tukabu; znamienne, że
większość złych magów stanowili mężczyźni — był to najwyraźniej specyficzny
sposób na wyrównanie „niedoboru" kobiet. To, że kuru występuje w związku z kanibalizmem uznano za pewnik w 1968 r. Dokładne wyjaśnienie specyfiki i podłoża
socjo-kulturowego rytuałów [za:] R. Rhodes, op. cit., s.12-15, 84-85 , materiały
Koła Naukowego Neurologii przy Klinice Neurologii Dorosłych Akademii Medycznej w Gdańsku [w:]
http://kolo-neuro.gumed.edu.pl/ [ 38 ] Inaczej
bezwład; brak koordynacji ruchów dowolnych bez uszkodzenia układu mięśniowego,
przyczyną są zmiany w obrębie ośrodkowego układu nerwowego. [ 39 ] Abazja — objaw kliniczny polegający na niemożności chodzenia, poruszania całością lub
częścią kończyny dolnej, niedowładzie lub całkowitym porażeniu spowodowanym
urazem. [ 40 ] Dokładnie:
toniczny odruch podeszwowy Hermana — odruch polegający na przetrwałym zgięciu
podeszwowym palców kończyny dolnej wywoływanym przez drażnienie podeszwy stopy.
Występuje w uszkodzeniach płata czołowego. [ 41 ] Ostatni
przypadek śmierci dziecka poniżej dziesięciu lat w plemieniu Fore miał miejsce w 1967 r. Wcześniej, w 1965 r., za pomocą szczepionek spreparowanych z zakażonej tkanki, udało się przenieść CJD na szympansy (Gajdusek został za to
uhonorowany w 1976 r. Nagrodą Nobla). Był to pierwszy niezbity dowód na to, że
niektóre choroby centralnego układu nerwowego mogą być zakaźne i pokonać barierę
gatunkową. Liczba zachorowań Fore na kuru systematycznie malała: od 1968 r. -
brak przypadków wśród dzieci w wieku 4-9 lat, od 1972 r. — brak zachorowań w grupie wiekowej dzieci od 10 do 14 lat, od 1973 r. — nie zarejestrowano
zachorowań wśród młodzieży od 15 do 19 roku życia. Równolegle śmiertelność
dorosłych spadła o dwie trzecie. [w:] R. Rhodes, op. cit.,s.104. « Biologia molekularna (Publikacja: 24-02-2011 Ostatnia zmiana: 25-02-2011)
str. 951 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||




 Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia.
Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia.
| [ Regulamin publikacji ] [ Bannery ] [ Mapa portalu ] [ Reklama ] [ Sklep ] [ Zarejestruj się ] [ Kontakt ] Racjonalista © Copyright 2000-2018 (e-mail: redakcja | administrator) | ||